
—— Estudi col·laboratiu dels CDC de Zhejiang, Macro & Micro-Test i els CDC de la Xina publicat a Frontiers in Cellular and Infection Microbiology
Resum de l'estudi
El maig de 2026, Frontiers in Cellular and Infection Microbiology (JCR Q1, IF ≈ 4.6) va publicar un article dirigit pel Centre Provincial de Control i Prevenció de Malalties de Zhejiang (CDC de Zhejiang), amb l'equip de bioinformàtica de Beijing Macro & Micro-Test Bio-Tech Co., Ltd. i l'Institut Nacional per al Control i la Prevenció de Malalties Transmissibles (CDC de la Xina) com a coautors. L'estudi es titula:
"Identificació i anàlisi filogenètica de set soques de Brucella abortus a Zhejiang, Xina."

Aquest estudi representa la primera anàlisi sistemàtica de traçabilitat filogenètica basada en el genoma complet de Brucella abortus (B. abortus) a la província de Zhejiang, Xina. L'equip va analitzar set aïllats recollits entre el 2015 i el 2025 (quatre soques d'origen humà i tres d'origen boví de Jinhua, Quzhou i Ningbo). Les troballes proporcionen evidència genòmica de l'origen i les vies de transmissió d'aquesta "espècie dominant del nord" en una regió epidèmica atípica del sud de la Xina oriental.
Antecedents i importància
La brucel·losi és una zoonosi causada per bacteris del gènere Brucella. Brucella abortus infecta principalment el bestiar boví, però també pot causar malalties en humans. A la Xina, la brucel·losi mostra una variació geogràfica marcada: la incidència més alta es produeix a les províncies del nord (per exemple, Mongòlia Interior, Shanxi, Heilongjiang). En canvi, les províncies del sud, inclosa Zhejiang, han estat històricament dominades per Brucella melitensis, amb molt pocs casos reportats de B. abortus. Aquesta disparitat regional fa que la caracterització genètica i la localització de l'origen de B. abortus a Zhejiang siguin una prioritat clau de salut pública.
Mètodes i principals troballes
L'equip de recerca va adoptar una estratègia múltiple que combina la biologia molecular i la bioinformàtica:
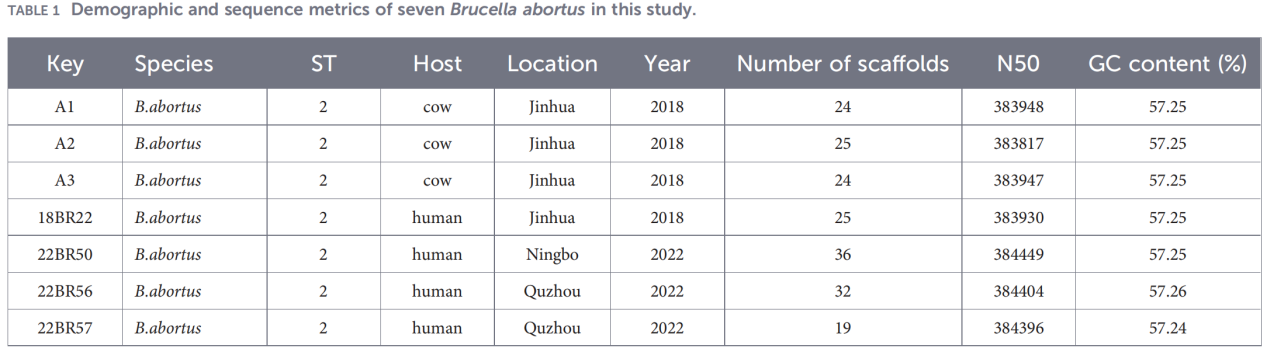
1.Identificació de patògens i tipificació bàsica
La PCR del gen BCSP-31 i l'AMOS-PCR van confirmar que els set aïllats eren B. abortus.
La tipificació de seqüències multilocus (MLST) basada en nou gens de manteniment va revelar que tots els aïllats pertanyien al tipus de seqüència ST2, cosa que indica una alta homogeneïtat genètica entre les soques de B. abortus circulants a Zhejiang.
2.Caracterització del genoma complet
La seqüenciació del genoma complet es va dur a terme a la plataforma Illumina NovaSeq. L'anàlisi d'identitat nucleotídica mitjana (ANI) va mostrar que els aïllats de Zhejiang compartien fins a un 99,99% de similitud amb la soca de referència B. abortus 544.
L'anàlisi pangenoma va revelar una població altament conservada: es van identificar 3.084 gens centrals, juntament amb només 10 gens de closca, i no es van detectar gens de nucli tou ni de núvol.
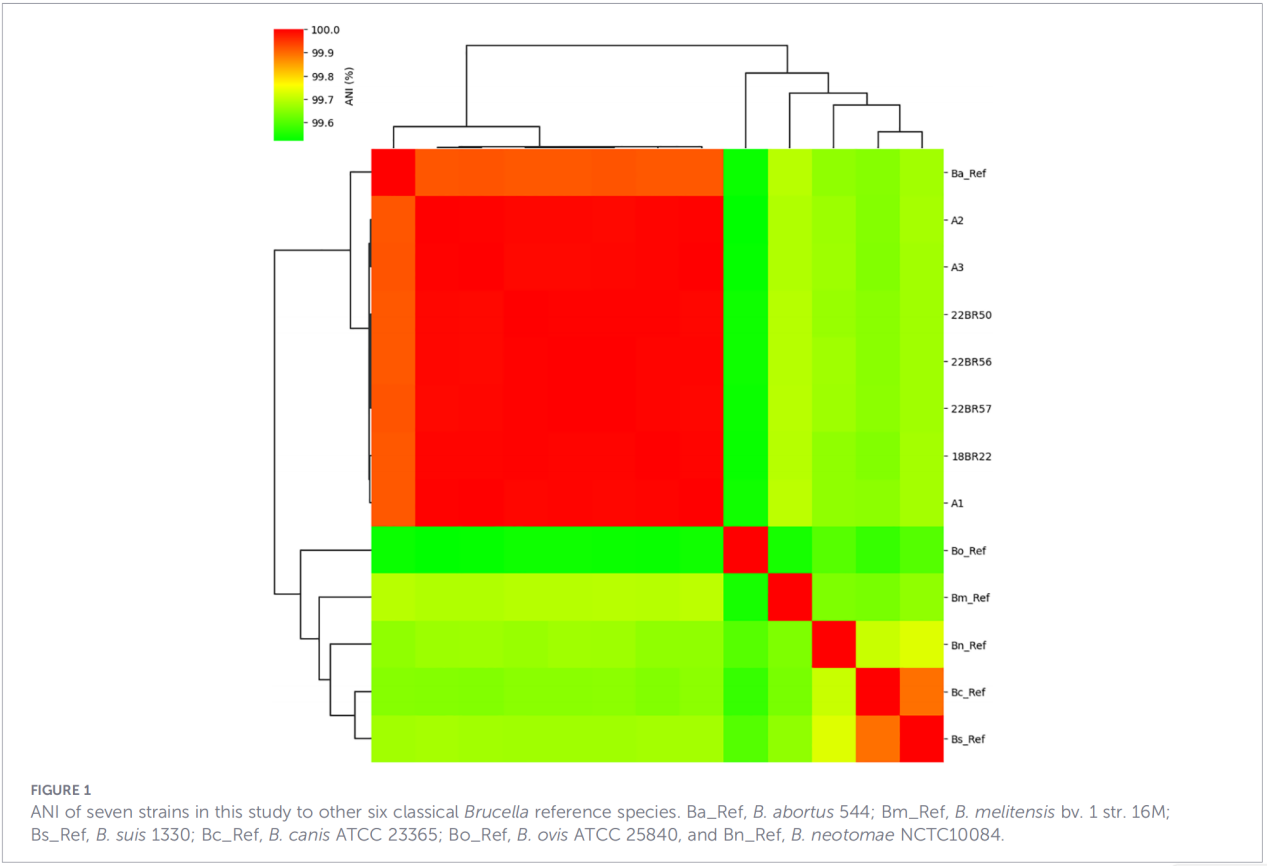
3.Perfils gènics de virulència i resistència antimicrobiana
Es van predir un total de 68 factors relacionats amb la virulència, que cobrien vies clàssiques com la biosíntesi de LPS, el sistema de secreció de T4SS i el sistema regulador de dos components BvrR-BvrS. Cal destacar que tots els aïllats no tenien els gens d'adhesina bmaA i btaF. L'anàlisi de gens de resistència només va detectar el gen mprF a la base de dades CARD, sense identificar altres determinants de resistència.
 4. Reconstrucció filogenètica i rastreig de transmissió
4. Reconstrucció filogenètica i rastreig de transmissió
L'anàlisi del polimorfisme d'un sol nucleòtid del genoma central (cgSNP) va situar els aïllats de Zhejiang en una posició específica de l'arbre filogenètic global. Els resultats van mostrar que les soques de Zhejiang formen un grup monofilètic juntament amb soques de Rússia, Mongòlia i diverses províncies del nord de la Xina (Ningxia, Heilongjiang, Mongòlia Interior, Hebei, Gansu, Pequín). Aquest grup es divideix a més en tres subclades diferents (Clade 1-3), cosa que suggereix múltiples esdeveniments d'introducció independents.
Conclusions i implicacions
Aquest estudi proporciona el primer conjunt de dades genòmiques d'alta precisió de B. abortus a la província de Zhejiang i arriba a diverses conclusions clau:
- Clear fons genètic– Les soques de B. abortus que circulen a Zhejiang pertanyen a ST2, estan genòmicament altament conservades i representen un llinatge típic de brucel·losi bovina.
2. Evidència de transmissió interregional– L'anàlisi filogenètica no avala l'existència d'un llinatge endèmic independent a Zhejiang. En canvi, les dades suggereixen fermament que aquestes soques es van originar del nord de la Xina i poden compartir un bagatge evolutiu comú amb soques de Rússia i Mongòlia. La presència de tres subclades implica múltiples esdeveniments d'introducció separats.
3. Implicacions per a la salut pública– Les troballes subratllen el valor de la vigilància genòmica de la brucel·losi, fins i tot en regions tradicionalment no endèmiques com ara Zhejiang. Tot i que el recompte de casos actual és baix, eines d'alta resolució com el cgSNP poden rastrejar eficaçment l'origen dels brots importats i proporcionar proves científiques per interrompre les cadenes de transmissió associades amb el transport interprovincial de bestiar.
Aquest treball no només omple un buit de recerca a la província de Zhejiang, sinó que també proporciona noves dades de referència per a la vigilància de patògens i l'avaluació del risc de brucel·losi a la regió del delta del riu Yangtze.
Informació del document:
Yang, Y., Shi, X., Chen, J., Wang, L., Wu, Z., Yao, W., … i Wu, B. (2026). Identificació i anàlisi filogenètica de set soques de Brucella abortus a Zhejiang, Xina. Frontiers in Cellular and Infection Microbiology, 16, 1758965.
Data de publicació: 10 de juny de 2026